深跨协总会工作汇报,行业协会地位重要性彰显

近期,全球疫情备受关注,很多外贸企业咨询口罩出口的详细要求及各国口罩准入条件,在此收集整理了海关总署及广东省市场监管局下属的广东省WTO/TBT通报咨询研究中心等单位发布的相关资料,在此分享给各位。
主要内容分为三部分:
1、口罩出口通关提示
2、口罩出口前准备
3、各国口罩准入条件
出口通关提示

报关前提条件:
收发货人注册编码(慈善机构可为临时编码),需办理无纸化通关法人卡
出口资质:
口罩出口对生产销售单位、境内发货人,除满足国内生产、市场流通资质需求外,中国海关无特殊资质要求。
出口申报要求:
1.商品归类:除特殊情况外,绝大部分口罩应归入税号63079000。
2.检验检疫:口罩为非法检产品,申报时检验检疫项目无需填报。根据我国政府与相关国家签订的政府间检验协议,对出口伊朗等少数几个国家的产品需按规定进行装运前检验。
3.关税征免:如出口物资为贸易性质,征免性质申报一般征税,征免方式申报照章征税;如为捐赠性质,境内发货人为贸易代理商、慈善机构等,征免性质可不填,征免方式申报全免。
4.禁限管理:目前商务部未对口罩设置贸易管制要求,中国海关也无针对防护物资的监管证件口岸验核要求。
5.申报规范:按照规范申报要求填写商品名称、成分含量;如物资非中国生产,原产国按照实际生产国填写。
出口退税:
口罩的出口退税率为13%。
中美关税排除加征:
美国企业可申请排除口罩进口加征关税,但是目前只有少数企业获准豁免。详见美国贸易代表办公室网站https://ustr.gov/。
快速通关保障:
物资出口申报如遇单窗等系统故障,可联系现场海关采取应急方式处置,或者拨打海关12360热线进行咨询。
出口前准备
以下内容是根据国内外相关政府机构、专业网站、新闻报道收集整理而成,仅供参考。具体内容以相关管理部门、国外官方机构要求为准。
明确口罩分类:
国外按照用途一般分为个人防护和医用两类口罩。
国内出口贸易企业需具备的资质和材料:
1.营业执照(经营范围有相关经营内容)。
2.企业生产许可证(生产企业)。
3.产品检验报告(生产企业)。
4.医疗器械注册证(非医用不需要)。
5.产品说明书(跟着产品提供)、标签(随附产品提供)。
6.产品批次/号(外包装)。
7.产品质量安全书或合格证(跟着产品提供)。
8.产品样品图片及外包装图片。
9.贸易公司须取得海关收发货人注册备案。
国内出口口罩生产企业资质证明:
生产个人防护或者工业用非医疗器械管理的普通口罩,有进出口权的企业,可自行直接出口。
生产属于医疗器械管理的口罩用于出口,中国海关不需要企业提供相关资质证明文件,但一般进口国会要求生产企业提供产品三证,以证明该进口的商品在中国已合法上市,具体如下:
1.营业执照(经营范围包含有医疗器械相关,非医疗级别的物品不需要)。
2.医疗器械产品备案证或者注册证。
3. 厂家检测报告。
生产企业有进出口权,可以自行出口,如没有进出口权,可以通过外贸代理进行出口销售。
内贸企业做出口需要取得的基本资质:
1. 向市场监管部门取得营业执照,增加经营范围“货物进出口、技术进出口、代理进出口”。
2. 向商务部门取得进出口权,可直接在商务部业务系统统一平台(http://iecms.mofcom.gov.cn/)申请,网上提交材料。
3. 向外汇管理局申请取得开设外汇账户许可。
4. 办理进出口货物收发货人海关注册登记。
各国口罩准入条件(产品准入条件)
1、必要资料:提单,箱单,发票。
2、个人防护口罩:必须取得美国 NIOSH检测认证,即National Institute for Occupational Safety and Health美国国家职业安全卫生研究所认证。
3、医用口罩:须取得美国FDA注册许可。
4、监管及出口须知:
据路透社报道,美国贸易代表处3月12日宣布,不对部分从中国进口的医药品加征关税。
这些医药品包括口罩、听诊器、血压计袖带等。
这一决定出于目前新冠疫情正在对美国健康医疗体系造成冲击。此前美国贸易代表处已将洗手液、医用手套等进口产品移出征税清单。
① FDA监管措施
FDA根据风险等级将医疗器械产品分为3个监管控制类别,涵盖近6000个产品代码(product code),根据不同的风险采取相应的监管力度。
不同类别医疗器械的监管措施:
类别 | 监管程度 | 是否需要申请510(k) |
I类 | 危险性小或基本无危险的产品,如非手术防护服,通过一般监管以保证其安全性和有效性。 | 多数产品可豁免510(k) |
II类 | 通过特殊监管来保证其安全性和有效性,包括特殊的标签要求、强制性指标、售后监控等,如手术用防护服。 | 多数产品需申请510(k),只有少数产品可豁免510(k) |
III类 | 最高风险类别产品,用于支持或维持人类生命、对预防人类健康损害起至关重要的产品,单独依靠一般监管和特殊监管不足以确保其安全性和有效性,往往需要上市前批准(PMA)。 | 需要申请比510(k)更严格的PMA |
FDA 510(k)又称上市前登记(Premarket Notification),是向FDA提交的上市前报告。FDA 510(k)的实质是证明器械的实质等同。
根据FDA的要求,少数I类和大部分II类医疗器械在美国上市前,至少需要提前90天递交510(k)申请,用于证明要销售的器械至少与已合法销售的器械(即等价器械)一样安全有效[21 CFR 807.92(a)(3)]。
在申请者收到FDA声明器械“实质性等同”(SE)的信件格式指令之前,不可在美国销售该器械。
510(k)的申请材料必须包括诸如产品代码、标签、器械技术特性的总结、测试结果等其他的资料。510(k)没有固定的表格或者模板,但是所有的信息必须符合《联邦法规》第21篇第807节中的要求。
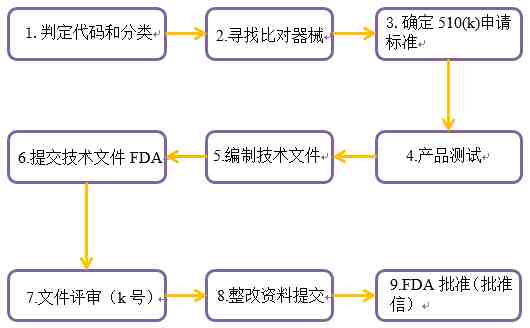
FDA 510(k)申请大致流程
如果FDA确定器械不是等价器械,申请者可以递交另一份含有新数据的510(k)文件,提出重新分类请求,或者递交上市前批准申批。
在FDA系统中对于口罩的分类代码有如下3个。其中一个是外科口罩,一个是儿科口罩,一个是带有抗菌/抗病毒介质的外科口罩。
三个类别的口罩都属于规则878.4040,分类都是Ⅱ类,都需要申请510(k)。
FDA不测试口罩,由申请者向FDA提供检测数据和产品宣称性能用于审核,检测内容包括颗粒过滤效率(PEF)、细菌过滤效率(BFE)、液体阻隔性、阻燃性等。510(k)申请周期较长,还有以下两种可选路径:
a)已经获得NIOSH批准的N95口罩可以直接注册
可以看出,如果制造商的N95口罩获得了NIOSH的批准,生物学测试、阻燃测试和血液穿透测试都通过了,那么则可以豁免510(k)的,可以直接进行工厂注册和器械列名。
b)获得持有510(k)的制造商的授权,作为其代工厂使用其510(k)批准号进行企业注册和器械列名。
申请方需要获得授权书,需要签署正式的质量协议,FDA会进行核实和抽查。如果使用未经许可的号码,将会导致产品召回的风险。
② NIOSH认证
医用口罩需要申请FDA 510(k)“上市前登记”。呼吸防护口罩则需要通过NIOSH认证,由NIOSH下属的NPPTL实验室实施认证。
口罩在出口美国之前,一般选择先做NIOSH认证,再做FDA认证,产品将更受美国市场欢迎。如医用N95口罩,需要既满足NIOSH对于N95口罩的要求,同时也要满足FDA相关标准。
NIOSH认证的流程如下:
1)提交申请书、技术资料(通常包括产品图纸、产品说明、质量体系文件、测试报告等)和样品;
2)美国实验室执行测试与评估,NIOSH颁发证书和标签;
3)生产商使用标签。
NIOSH规定单独的过滤式呼吸防护口罩上必须具有以下标识:
1)NIOSH认可的批准持有人/制造商名称,注册商标或申请人/批准持有人的企业名称缩写。如果适用,批准持有人对呼吸防护口罩进行私有标记的实体名称可以代替NIOSH认可的批准持有人的业务名称,注册商标或批准持有人的业务名称的缩写;
2)NIOSH的大写或NIOSH徽标;
3)NIOSH测试和认证批准号,例如TC-84A-XXXX;
4)NIOSH过滤特性和过滤效率等级,例如N95、N99、N100、R95、P95、P99、P100(目前NIOSH批准的过滤式呼吸防护口罩的七种类型);
5)型号或零件号:批准持有人的呼吸器型号或零件号,由一系列数字或字母数字标记表示,例如8577或8577A。
NIOSH建议还包括批号和/或生产日期,但这不是必需的。
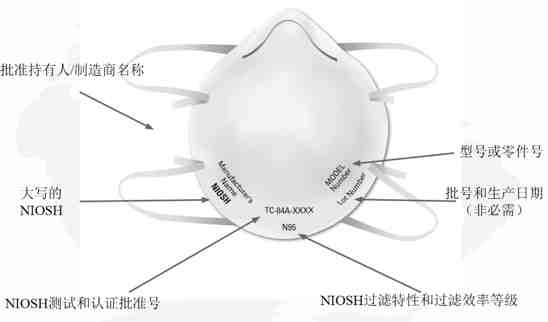
NIOSH过滤式呼吸防护口罩标识
1、必要资料:提单,箱单,发票。
2、个人防护口罩:个人防护口罩的欧盟标准是EN149,按照标准将口罩分为FFP1/FFP2和FFP3三个类别。所有出口欧盟的口罩必须获得CE认证证书。CE认证是欧盟实行的强制性产品安全认证制度,目的是为了保障欧盟国家人民的生命财产安全。
3、医用口罩:医用口罩对应的欧盟标准是EN14683。产品在欧盟销售需要出具欧盟自由销售证书 Free Sale Certificate,有了CE标志并进行了相关指令中要求的欧盟注册后,中国的制造商出口欧盟不需要自由销售证书。
4、监管及出口须知:
① 呼吸防护口罩
呼吸防护口罩需要满足法规(EU) 2016/425的要求,防护口罩属于其中复杂设计的产品。出口欧盟需要授权的公告机构(NB)进行认证并颁发证书,认证流程为:
a)提供申请表、产品实物图片及说明书;
b)准备产品型式试验报告。(依据EN 149检测);
c)技术文件评审(由发证机构评审);
d)工厂质量体系审查(由发证机构评审工厂体系资料);
e)公告机构颁发CE证书。
② 医用口罩
医用口罩产品可分为无菌或非无菌状态,其认证模式不一样。
非无菌状态认证:
a)编制技术文件(TCF);
b)提供测试报告(依据EN 14683要求检测,或提供熔喷布性能测试报告和无纺布生物学测试报告);
c)编制产品符合性声明(DoC);
d)指定欧盟授权代表并完成欧洲注册。
无菌状态认证:
a)灭菌验证;
b)建立ISO 13485医疗器械质量管理体系;
c)编制技术文件(TCF);
d)提供测试报告(依据EN 14683要求检测,主要提供细菌过滤效率、呼吸阻力、防溅阻力及灭菌验证报告等);
e)公告机构(NB)审核;
f)获ISO 13485证书和CE证书;
g)指定欧盟授权代表并完成欧洲注册。
从目前整体情况来看,如果之前没有获得公告机构的CE证书,现在临时申请周期比较长。因此,企业可考虑出口非无菌医用口罩。但是非无菌医用口罩并不是对生产环境完全无限制,EN 14683对于产品的初始污染菌要求不大于30 cfu/g。
1、必要资料:提单,箱单,发票,日本国外的制造商必须向PMDA注册制造商信息。
2、口罩包装要求:
包装上印有ウィルスカット(中文翻译:病毒拦截)99%的字样
PFE:0.1um微粒子颗粒过滤效率
BFE:细菌过滤率
VFE:病毒过滤率
3、口罩品质标准:
1) 医用防护口罩:符合中国GB 19083-2010 强制性标准,过滤效率≥95%(使用非油性颗粒物测试)。
2)N95口罩:美国NIOSH认证,非油性颗粒物过滤效率≥95%。
3)KN95口罩:符合中国GB 2626 强制性标准,非油性颗粒物过滤效率≥95%。
4、监管及出口须知:
由于非工业用口罩没有关于性能的鉴定规范,会造成口罩的标识和广告的内容差异较大而给消费者带来巨大误解的情况。
为此,日本口罩行业协会于2006年1月制定并实施了关于口罩的“标识和广告自愿性标准”,并呼吁所有协会会员的口罩制造商从保护消费者的角度出发,履行其社会责任。
日本全国口罩行业协会的会员标志如下:

JHPIA对口罩的标识和广告的规定,不得在口罩的容器、外包装以及广告上声称以下内容:
①声称具有医疗用品方面的功效和效果,声称具有医药品、药妆、化妆品、医疗器械的功效和效果;
②缺乏依据声称口罩滤料的收集效率数值的标识(但是,在有依据的情况下,可在标识出检测方法或者检测机关的前提下标识该数值,收集效率最高为99%)。
统一框内标识格式如下:
产品名称 | 填写「マスク」(编者注:口罩)。此外,在「マスク」后面用括弧注明(商品名〇〇〇)。 |
过滤物质 | 注明“風邪・花粉・ホコリ”(编者注:感冒、花粉、灰尘)中哪一个物质。 |
材质 | 注明口罩本身、耳挂、滤材所使用的材质 |
抗菌剂名称 | 如使用了抗菌剂,应在框内设一栏注明抗菌剂的成分。 |
数量 | 注明口罩的数量。 |
企业名 | 如制造商与销售商相同,则注明制造和销售商的名称。 如制造商与销售商不同,则注明对消费者负责一方的名称。 |
包装材料的材质 | 注明包装材料的材质。 |
1、必要资料:提单,箱单,发票,韩国进口商营业执照。
2、个人防护口罩标准:KF (Korean filter) 系列分为KF80、KF94、KF99
3、执行标准规范:MFDS Notice No. 2015-69
韩国医疗器械准入的法规门槛,基本分类为I、II、III、IV类,持证为韩国公司(License holder),韩国收货人需要到韩国药监局Korea Pharmaceutical Traders Association. 提前备案进口资质(没有不行)网址:www.kpta.or.kr。
4、监管及出口须知:
根据《药品事务法》,医药辅品是指与疾病的治疗和预防有关的产品,并由食品药品安全局局长指定的,用于治疗,减少,治疗或预防人类或动物疾病的纺织品和橡胶制品、对人体无害或不直接影响人体的产品以及用于灭菌,杀虫剂和类似目的的产品以预防传染病的产品。
在韩国,这一分类下的物品有:口罩(手术用、卫生保健用)、用于保护、处置患处的产品(如:眼罩、绷带、纱布等)、卫生巾、口腔卫生用品、直接用于人体外部消毒剂(如洗手液)等。
医药辅品上市/进口前需先向食品医药品安全评估院或地方食品医药品安全厅进行申请许可。
根据《医药辅品的批准、通知、评价规定》,在初次申请卫生口罩的认可时,应提供测试结果作为支持数据。指定准药物范围-执法20211001食品药品安全部公告2019-86-2019年9月30日部分修订额定值的泄漏率不得大于25.0%,KF94额定值的泄漏率不得大于11.0%,KF99额定值的泄漏率不得大于5.0%。口罩的额定值(如:KF80、KF90、K99)应在产品名称括号中注明。
卫生口罩需并在成品上注明“按下列试验方法进行试验时,各单项测量值不应小于OOO%。”并在描述面部吸入阻力试验测试方法时说明以下细节:“将标准头部模型放置在面部区域后进行试验。使用六个卫生口罩时,三个应与提交的一样,另外三个应在温度38±2.5℃和湿度85±5%RH下无人看管24±1小时,然后用作试样。当空气以每分钟30升的连续流量喷过面部区域时,应测量水柱(mmH2O),并详细说明试验方法的其他细节。”
根据2014年9月4日的补充规定2014-153号,原本作为防尘口罩或防病口罩的产品,被允许作为公共卫生口罩。
如果得到韩国食药厅许可,口罩会在包装上标上 “의약외품(医药辅品)”。“수술용(手术用)”和“보건용(卫生保健用)”都属于医药辅品。
根据韩国海关官网2020年3月5日信息,口罩进口清关流程如下:
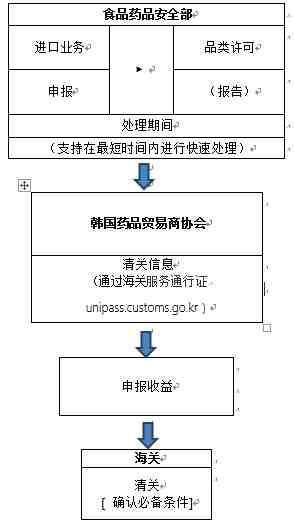
目前食品药品安全部正在进行快速许可,许可审查部门的联系方式如下:(联系以获取进口业务报关单和产品授权书,并迅速处理进口要求。 )
部门 | 联络方式 |
食品药品安全部 融合创新产品支持小组批准总队 | 043-719-2333 |
首尔地方食药厅医药品安全管理科 | 02-2640-1413 |
釜山地方食药厅医疗产品安全科 | 051-602-6187 |
京仁地方食药厅医疗产品安全科 | 02-2110-8097、8072 |
大邱地方食药厅医疗产品安全科 | 053-589-2757 |
光州地方食药厅医疗产品安全科 | 062-602-1541,1455 |
大田地方食药厅医疗产品安全科 | 042-480-8768 |
1、必要资料:提单,箱单,发票。
2、须通过澳洲的TGA注册,符合标准规范:AS/NZS 1716:2012,此规范是澳大利亚和新西兰的呼吸保护装置标准。
TGA 是Therapeutic Goods Administration的简写,全称是治疗商品管理局,它是澳大利亚的治疗商品(包括药物、医疗器械、基因科技和血液制品)的监督机构。澳大利亚对医疗器械分为I类,Is and Im, IIa, IIb, III类,产品的分类几乎和欧盟分类一致,如果产品已经获得CE标志,则产品类别可以按照CE分类。
3、监管及出口须知:
澳洲的医用口罩按照I类管理,需要在TGA进行备案之后销售。在澳洲的备案需要由澳洲当地的SPONSOR来完成,其合规流程为:
① 指定SPONSOR;
只有通过澳大利亚的代理人才能够提出申请,代理人这里有一个专有名词叫“Sponsor”,简单来说就是进口商。
② 完成技术文档;
低风险的I类器械没有强制性质量体系和上市前评价的明确要求, 但要求制造商提供相关文件证明其安全有效性。
③ 提交TGA进行备案;
④ 获得证书。
特别提醒:澳大利亚已与欧盟达成互认协议。这意味着,合格评定证书由TGA颁发的也被欧盟认可,TGA也认可欧盟CE认证。已获CE认证的用户,可提交CE证书及相关资料,获得TGA证书。
资料来源:
1、微信公众号:12360海关热线,《促外贸稳增长-海关技术性贸易措施指南(口罩出口篇)》,2020.03.17
2、广东省WTO/TBT通报咨询研究中心,《防护服等产品法规标准出口知识介绍》,2020.03.16
提示:
以上内容是根据国内外相关政府机构、专业网站、新闻报道收集整理而成,仅供参考。具体内容以相关管理部门、国外官方机构要求为准。
———— e n d ————

我们建了一个亚马逊卖家交流群,里面不乏很多大卖家。
现在扫码回复“ 加群 ”,拉你进群。





热门文章
*30分钟更新一次